Banishing Bad Hair Days since 1997!™
Search HairBoutique
Articles
Blogs
Celebs
Hair Loss
HairTalk®
Ask Karen™
Gallery
Glossary
Forum Home
>
Hip Hop
>
Hip Hop Talk Archives
Forum Options
New Posts
FAQ
Search
Register
Login
Hip Hop Talk Archives
New Topic
Show Topics
Any Date
1 second
ago at 5:03pm
Yesterday
Last 2 Days
Last Week
Last Month
Last Two Months
Last Six Months
Last Year
Page
<
1
2
3
4
5
>
Topics
/
Topic Starter
Rating
Replies
Views
Last Post
Forum Topics
cassidy a murderer?
By
blackwallstreet
, June 19 2005 at 8:28pm
1
594
By
The Man
June 28 2005 at 11:06am
Lil� Kim?
By
Jesuschrist#1
, July 07 2005 at 10:40pm
0
520
By
Jesuschrist#1
July 07 2005 at 10:40pm
Chris Brown - Da boy can sing
By
JHood
, July 08 2005 at 9:40pm
1
821
By
pmp4lfjmike
July 08 2005 at 11:43pm
battle
By
jayrip2009
, July 11 2005 at 11:29pm
12
1603
By
JHood
July 14 2005 at 5:05pm
BATTLE
By
jayrip2009
, July 12 2005 at 2:45am
12
1683
By
pmp4lfjmike
July 12 2005 at 8:45pm
RULES
By
blackwallstreet
, July 14 2005 at 1:10pm
1
527
By
blackwallstreet
July 15 2005 at 8:46am
Who won
By
Rence119
, July 17 2005 at 11:19am
4
721
By
Rence119
July 21 2005 at 7:58pm
best freestyler
By
blackwallstreet
, July 22 2005 at 12:08pm
18
2369
By
Rence119
July 27 2005 at 7:53am
check dis out
By
Bowizzle
, July 24 2005 at 5:21pm
7
998
By
Rence119
July 26 2005 at 8:05pm
Rence,Black vsPmp and bowizzle
By
Rence119
, July 26 2005 at 3:56pm
1
2
3
49
4762
By
Rence119
July 28 2005 at 10:36pm
black is a pussy
By
Bowizzle
, July 29 2005 at 9:31pm
1
547
By
Rence119
July 30 2005 at 10:57am
poll
By
Bowizzle
, July 29 2005 at 10:17pm
0
441
By
Bowizzle
July 29 2005 at 10:17pm
gotwaves&rence vs pmp&bowizzle
By
gotwaves
, July 29 2005 at 11:32pm
1
2
3
51
4086
By
Rence119
August 03 2005 at 3:36pm
bowizzle and pmp vs gotnowaves and lil j
By
Bowizzle
, August 02 2005 at 6:49pm
18
2226
By
Rence119
August 04 2005 at 3:54pm
tag team
By
Lil t
, August 04 2005 at 8:29am
1
2
21
2687
By
Rence119
August 23 2005 at 11:50am
battle
By
Lil t
, August 04 2005 at 12:55pm
1
2
30
3691
By
LIL J
August 14 2005 at 8:46pm
BLACK IZ BACK
By
blackwallstreet
, August 08 2005 at 2:14pm
1
2
21
3042
By
Rence119
August 14 2005 at 9:40am
Listen Lil J
By
Rence119
, August 13 2005 at 4:00pm
11
1163
By
LIL J
August 15 2005 at 1:34pm
Mase & G Unit
By
G Slim
, August 20 2005 at 4:50pm
1
2
29
4007
By
Rence119
September 23 2005 at 10:03pm
lets start a tourament
By
Bowizzle
, August 20 2005 at 7:09pm
13
1562
By
Rence119
August 30 2005 at 9:52pm
The Diss to blackstreet
By
pmp4lfjmike
, August 20 2005 at 7:37pm
14
1756
By
Rence119
August 30 2005 at 9:53pm
Im Back
By
Rence119
, August 23 2005 at 11:54am
2
529
By
Rence119
August 24 2005 at 9:26pm
VMA awards
By
Rence119
, August 31 2005 at 10:32am
1
2
25
3134
By
Rence119
September 13 2005 at 7:45pm
The Last Diss to Wallstreet and last rap
By
pmp4lfjmike
, August 31 2005 at 12:38pm
1
2
3
4
5
88
9145
By
Rence119
September 13 2005 at 7:42pm
All Due Respects
By
blackwallstreet
, September 02 2005 at 2:37pm
10
1385
By
Rence119
September 18 2005 at 7:07pm
off tha dome
By
blackwallstreet
, September 02 2005 at 5:42pm
6
1025
By
Rence119
September 05 2005 at 12:02am
where my niccas at
By
Rence119
, September 04 2005 at 9:23am
13
1326
By
Rence119
September 09 2005 at 2:13pm
is it true?
By
blackwallstreet
, October 01 2005 at 6:29pm
6
1147
By
Rence119
October 09 2005 at 8:16pm
Beanie Sigal signing with Gunit
By
G Slim
, October 02 2005 at 3:33pm
7
1340
By
Rence119
October 07 2005 at 7:22pm
RRS BABY
By
STACKBUNDLES
, June 08 2005 at 12:10pm
2
628
By
STACKBUNDLES
June 19 2005 at 2:12pm
New Topic
Page
<
1
2
3
4
5
>
Forum Jump
-- Select Forum --
HairBoutique.com News
Introductions
Buy/Sell/Swap Hair
Forum How-To's
- Andrew's Corner
Hook-ups
Announcements
Beauty Job Board
Book Reviews
- Book Exchange
Feedback/Suggestions
Friendship Forum
Personals
Work, Work, Work
Help Wanted/Situations Wanted
African American Hair
Braid Talk
African American Hair & Related History
African American Men's HairTalk®
Celebrity Hair Talk
Celebrity Hair & HairstylesTalk Archives
Reality Show Talk
Celebrity Juice
Aromatherapy
Beauty News
Cosmetics
Cosmetic Surgery
Diet Days
Nails
Perfumery
Professional Ponderings
Skin Care
Curly Hair
Hot Iron Comments & Feedback
Curly Haired Celebs
Curly Hair In The News
Curly Hair Typing
Curly Hair - Type 2
Curly Hair - Type 3
Curly Hair - Type 4
Curly Hair Styling Experts
Curly Hair Product Reviews
Curl Related Book Reviews
Straight Talk
A Passion For Fashion
Handbags
Shoes, shoes & shoes
Earrings Galore
Prom Hairstyles
Prom & Prom Hairstyles 2011-2012
Up, Down & Formal Hairstyles
Wedding Hairstyles
Prom Stories
Prom Dresses
Prom Articles & Links
Buy/Sell Prom Gowns & Accessories
Eyelash Extensions
Hair Extensions
Extension Links & Trades
Group Orders & Related
Hair Extension Faqs & Facts
Looking For Hair Extension Expert
Shrink*** & Related Topics
Off Topics
Lace Front Wig Talk
Off Topics
Lace Front Wigs Links & Trades
Archives & Locked Threads
Hair Journals
Hair Polls
Emo Hair
Twilight & Vampire Hair
Teen Hair & Hairstyles
Asian Hair
Astrology & Hair
Bloopers
Dreadlocks
Fine Hair
Food For Hair
General Hair Talk
Hair & Alternative Therapies
Hair Color
Hair Loss
Hair News
Hair Politics
Senior Strands
Short Hair
Hip Hop - You Against Me - Battle Forum
Hip Hop Hang Out Zone
Hip Hop Happenings
Hip Hop News
Poetry / The Spoken Word
Hip Hop Fashion
At The Movies
VIP Team
Hip Hop Talk Archives
Long Hair Support
Long Hair Care Experts
Men's Hair
Men's Hair Loss
Men's Grooming Topics
Men's Fashion Accessories
Men's Fashion
Travel Talk
Sewing, Knitting, Quilting & Related
Entertainment Talk
Dogs & Cats & Pet Talk
Food Groupies
Fun Zone
Hair Haiku & Poetry
Beauty & Hair Care Recipes
Swap, Share, Trade, Etc.,
Product Finder
Product Reviews
Salon Finder
Professional HairDressers
Professional Schools & Education
Beauty Shows
Nail Techs
360 Waves HowTos
Men's 360 Waves - Part II
- 180 Vet Methods
- 360 Wave Vets - Private Club
Getting Started
360 Product Reviews
Men's 360 Waves Archives
360 Wave Gallery
360 Battle Forum
360 Off Topics & Hang Out
The Ladies
Cars
Gaming Zone
720 Waves
TV Talk
The Soaps
TV Show News & Scoops
Ugly Betty
Brothers & Sisters
Heroes
Baby Hair Talk
Pregnancy Hair & Beauty Tips
Women's Health Talk
Hair Talk Archive
Hair Vitamins & Growth Formulas
Other Marketplace Products
- Karen's Corner
Favorite Recipes
Restaurant Reviews
General Food Tips & Tricks
Reality Foodie Shows
Making Money Tips
News Items
Forum Permissions
You
cannot
post new topics in this forum
You
cannot
reply to topics in this forum
You
cannot
delete your posts in this forum
You
cannot
edit your posts in this forum
You
cannot
create polls in this forum
You
cannot
vote in polls in this forum
Hair Tips: Fake Bob Hairstyles
Rossano Ferretti's Invisible Scissors Hair-Cutting Method
A Safer, Smarter Weight Loss Formula Than Ozempic Or GLP-1
Is My Hair Stylist Overcharging Me For My Hair Extensions?
Fatten Fine Hair With Yogurt And Cocoa
5-Minute Faster Hair Growth
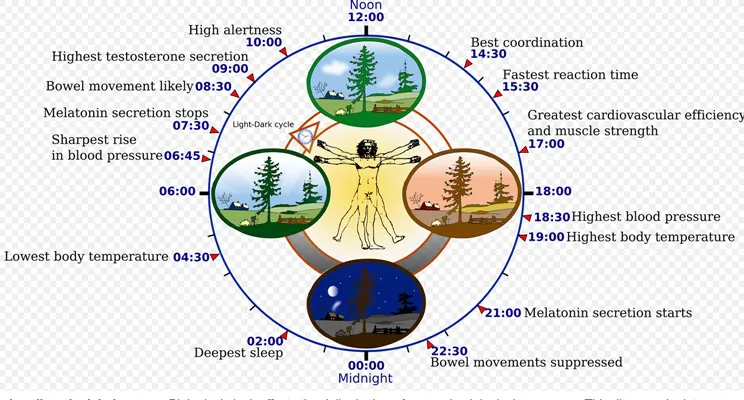
Hair Growth And Circadian Rhythms
Fast Hair Growth Lies
Memories Of Robert Hallowell's Best Hair Recipes
Amy Poehler's Classic Bob Hairstyle Through The Years
Kirsten Dunst's Career & Glam Hair Are Back In Action
You Can Use This Hair Milk Even If You’re Lactose Intolerant
Maggie Rogers' Rockin Hairstyle
Maren Morris' W Curtain Bangs
What Is The Wolf Hair Cut?
What I Learned as the Only White Student at a Black Hair Academy
Hair Myths: Don’t Believe Everything You Hear
Will Jennifer Aniston's Endorsement Hurt Hair Care Line?
10 Commandments for a Great Haircut
Why Dr. Oz Tells Overweight Men - Lose 35 Pounds For 1 Inch Gain
Copyright 1997-2025,
hairboutique.com
, All Rights Reserved.
Terms of Service
,
Privacy Statement
,
Advertise
,
Contact Us
,
Press
,
Disclaimer








 Hip Hop Talk Archives
Hip Hop Talk Archives